In our previous post on bioinformatics overview, we were able to list a number of applications of the methods of computer science as they apply to the collection, storage, organization, analysis, or Biological data visualization. This article will talk about how to present, structure data as well as how to process DNA data. Along with that, the article also provides a small example of how to classify a genome family using a simple machine learning method.
What is DNA
DNA (Deoxyribonucleic Acid), also known as DNA (acide désoxyribonucléique) is a molecule that contains biological instructions that make up the characteristics of each species. DNA, along with the genetic information it contains, is passed from adult organisms to their offspring during reproduction.
The human genome has about 20,000-25,0000 genes, which are gathered inside the chromosomes (chromosomes) located in the nucleus of a cell. The human genome consists of about 3 billion base pair molecules, the human DNA pairs are made up of four types of nucleotides including “A”, “C”, “G”, “T”. We all have separate genomes but still share many of the same parts.
For DNA data, machine learning techniques can be applied ddeer:
- Capture the dependence of the data
- Inference and discovery of new biological hypotheses
So in this article, we will learn how to interpret the structure of DNA and how machine learning algorithms can be used to build predictive models on DNA sequence data.
How the DNA is represented
Most DNA molecules are made up of two evenly twisted biopolymers around an imaginary axis forming a double helix.

Illustrations from https://biologydictionary.net/double-helix/
These two DNA strands are called polynucleotides because their composition is composed of nucleotide monomers. Each nucleotide is made up of one of four nitrogen-containing nucleobases — either cytosine (C), guanine (G), adenine (A), or thymine (T) — bound to the sugar deoxyribose and a phosphate group. The nucleotides are linked together into a DNA circuit by covalent bonding between the sugar molecule of the nucleotide and the phosphate group of the next nucleotide, forming a solid rotating sugar-phosphate “backbone”.
The order or sequence of nucleobases determines which biological instructions are contained in a DNA sequence. For example, a specific region on chromosome 15 is important in determining eye color.
How Python treats DNA data
We all know that, Python has a lot of libraries that support data processing and visualization, part of which is biological data. Two of the libraries may be mentioned is Biopython and squiggle
- Biopython is a set of python modules that provide functions to handle the operations of DNA, RNA, and protein sequences such as reverse addition of DNA sequences, finding modules in protein chains, etc.It provides a lot of work. parser to read all major genetic databases like GenBank, SwissPort, FASTA, etc,
- Squiggle is a software tool that automatically generates web-based two-dimensional graphic representations of raw DNA sequences. Squiggle has implemented several previously published sequence visualization algorithms and introduces new visualization methods designed to maximize user usability.
To install the above two libraries, starting with the Jupyter Notebook environment we use the following commands:
1 2 3 | !pip install biopython !pip install Squiggle |
Genetic data is usually stored in a number of formats, one of which is FASTA. When we open an FASTA file, we can see the content will be similar to this:
1 2 3 4 5 6 7 | >HSBGPG Human gene for bone gla protein (BGP) GGCAGATTCCCCCTAGACCCGCCCGCACCATGGTCAGGCATGCCCCTCCTCATCGCTGGGCACAGCCCAGAGGGT ATAAACAGTGCTGGAGGCTGGCGGGGCAGGCCAGCTGAGTCCTGAGCAGCAGCCCAGCGCAGCCACCGAGACACC ATGAGAGCCCTCACACTCCTCGCCCTATTGGCCCTGGCCGCACTTTGCATCGCTGGCCAGGCAGGTGAGTGCCCC CACCTCCCCTCAGGCCGCATTGCAGTGGGGGCTGAGAGGAGGAAGCACCATGGCCCACCTCTTCTCACCCCTTTG ...... |
To see the full content of the above example file, anyone can see http://www.cbs.dtu.dk/services/NetGene2/fasta.php . The entire contents of the FASTA file are in bold on the web page above. Copy that part and save it in .fa format like normal text file such as example.fa, they can use that file as data for
Squiggleexample.
To use matplotlib well with Jupyter Notebook we use eternal magic command
1 2 | <span class="token operator">%</span> matplotlib inline |
Next, with the Biopython library we can read the information of the data file with the following commands:
1 2 3 4 5 6 7 | <span class="token keyword">from</span> Bio <span class="token keyword">import</span> SeqIO <span class="token keyword">for</span> sequence <span class="token keyword">in</span> SeqIO <span class="token punctuation">.</span> parse <span class="token punctuation">(</span> <span class="token string">'./example.fa'</span> <span class="token punctuation">,</span> <span class="token string">'fasta'</span> <span class="token punctuation">)</span> <span class="token punctuation">:</span> <span class="token keyword">print</span> <span class="token punctuation">(</span> sequence <span class="token punctuation">.</span> <span class="token builtin">id</span> <span class="token punctuation">)</span> <span class="token keyword">print</span> <span class="token punctuation">(</span> sequence <span class="token punctuation">.</span> seq <span class="token punctuation">)</span> <span class="token keyword">print</span> <span class="token punctuation">(</span> <span class="token builtin">len</span> <span class="token punctuation">(</span> sequence <span class="token punctuation">)</span> <span class="token punctuation">)</span> |
The results are as follows:
1 2 3 4 5 6 7 | HSBGPG GGCAGATTCCCCCTAGACCCGCCCGCACCATGGTCAGGCATGCCCCTCCTCATCGCTGGGCACAGCCCAGAGGGTATAAACAGTGCTGGAGGCTGGCGGGGCAGGCCAGCTGAGTCCTGAGCAGCAGCCCAGCGCAGCCACCGAGACACCATGAGAGCCCTCACACTCCTCGCCCTATTGGCCCTGGCCGCACTTTGCATCGCTGGCCAGGCAGGTGAGTGCCCCCACCTCCCCTCAGGCCGCATTGCAGTGGGGGCTGAGAGGAGGAAGCACCATGGCCCACCTCTTCTCACCCCTTTGGCTGGCAGTCCCTTTGCAGTCTAACCACCTTGTTGCAGGCTCAATCCATTTGCCCCAGCTCTGCCCTTGCAGAGGGAGAGGAGGGAAGAGCAAGCTGCCCGAGACGCAGGGGAAGGAGGATGAGGGCCCTGGGGATGAGCTGGGGTGAACCAGGCTCCCTTTCCTTTGCAGGTGCGAAGCCCAGCGGTGCAGAGTCCAGCAAAGGTGCAGGTATGAGGATGGACCTGATGGGTTCCTGGACCCTCCCCTCTCACCCTGGTCCCTCAGTCTCATTCCCCCACTCCTGCCACCTCCTGTCTGGCCATCAGGAAGGCCAGCCTGCTCCCCACCTGATCCTCCCAAACCCAGAGCCACCTGATGCCTGCCCCTCTGCTCCACAGCCTTTGTGTCCAAGCAGGAGGGCAGCGAGGTAGTGAAGAGACCCAGGCGCTACCTGTATCAATGGCTGGGGTGAGAGAAAAGGCAGAGCTGGGCCAAGGCCCTGCCTCTCCGGGATGGTCTGTGGGGGAGCTGCAGCAGGGAGTGGCCTCTCTGGGTTGTGGTGGGGGTACAGGCAGCCTGCCCTGGTGGGCACCCTGGAGCCCCATGTGTAGGGAGAGGAGGGATGGGCATTTTGCACGGGGGCTGATGCCACCACGTCGGGTGTCTCAGAGCCCCAGTCCCCTACCCGGATCCCCTGGAGCCCAGGAGGGAGGTGTGTGAGCTCAATCCGGACTGTGACGAGTTGGCTGACCACATCGGCTTTCAGGAGGCCTATCGGCGCTTCTACGGCCCGGTCTAGGGTGTCGCTCTGCTGGCCTGGCCGGCAACCCCAGTTCTGCTCCTCTCCAGGCACCCTTCTTTCCTCTTCCCCTTGCCCTTGCCCTGACCTCCCAGCCCTATGGATGTGGGGTCCCCATCATCCCAGCTGCTCCCAAATAAACTCCAGAAG <span class="token number">1231</span> HSGLTH1 CCACTGCACTCACCGCACCCGGCCAATTTTTGTGTTTTTAGTAGAGACTAAATACCATATAGTGAACACCTAAGACGGGGGGCCTTGGATCCAGGGCGATTCAGAGGGCCCCGGTCGGAGCTGTCGGAGATTGAGCGCGCGCGGTCCCGGGATCTCCGACGAGGCCCTGGACCCCCGGGCGGCGAAGCTGCGGCGCGGCGCCCCCTGGAGGCCGCGGGACCCCTGGCCGGTCCGCGCAGGCGCAGCGGGGTCGCAGGGCGCGGCGGGTTCCAGCGCGGGGATGGCGCTGTCCGCGGAGGACCGGGCGCTGGTGCGCGCCCTGTGGAAGAAGCTGGGCAGCAACGTCGGCGTCTACACGACAGAGGCCCTGGAAAGGTGCGGCAGGCTGGGCGCCCCCGCCCCCAGGGGCCCTCCCTCCCCAAGCCCCCCGGACGCGCCTCACCCACGTTCCTCTCGCAGGACCTTCCTGGCTTTCCCCGCCACGAAGACCTACTTCTCCCACCTGGACCTGAGCCCCGGCTCCTCACAAGTCAGAGCCCACGGCCAGAAGGTGGCGGACGCGCTGAGCCTCGCCGTGGAGCGCCTGGACGACCTACCCCACGCGCTGTCCGCGCTGAGCCACCTGCACGCGTGCCAGCTGCGAGTGGACCCGGCCAGCTTCCAGGTGAGCGGCTGCCGTGCTGGGCCCCTGTCCCCGGGAGGGCCCCGGCGGGGTGGGTGCGGGGGGCGTGCGGGGCGGGTGCAGGCGAGTGAGCCTTGAGCGCTCGCCGCAGCTCCTGGGCCACTGCCTGCTGGTAACCCTCGCCCGGCACTACCCCGGAGACTTCAGCCCCGCGCTGCAGGCGTCGCTGGACAAGTTCCTGAGCCACGTTATCTCGGCGCTGGTTTCCGAGTACCGCTGAACTGTGGGTGGGTGGCCGCGGGATCCCCAGGCGACCTTCCCCGTGTTTGAGTAAAGCCTCTCCCAGGAGCAGCCTTCTTGCCGTGCTCTCTCGAGGTCAGGACGCGAGAGGAAGGCGC <span class="token number">1020</span> |
Of course besides just printing it out we can use the Squiggle library mentioned above to visualize the data. Using the command below, Squiggle will open a data visualization web page on which we can move, zoom, and interact with the data.
1 2 | !squiggle <span class="token punctuation">.</span> <span class="token operator">/</span> example <span class="token punctuation">.</span> fa |
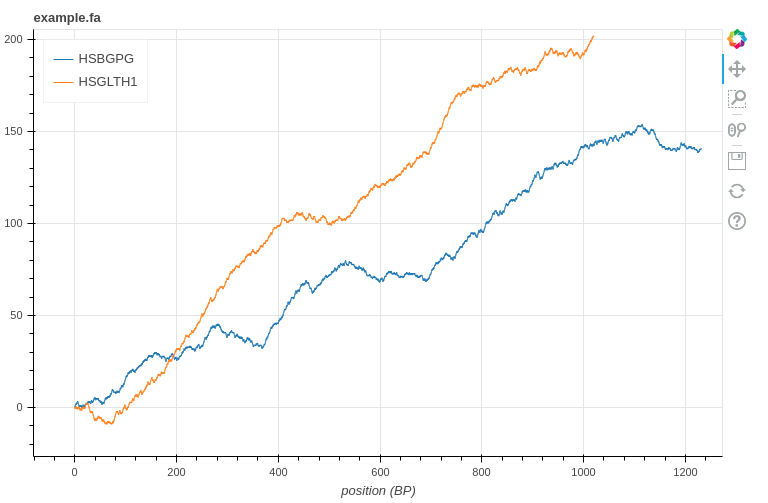
There are also many
Squiggleoptions that we can learn more about at the library’s documentation page.
Predict genetic families based on machine learning methods
A genomic family is a set of several similar genes formed by duplicating an original gene that generally has a similar biochemical function. Genes are classified into families based on the shared sequence of nucleotides or proteins.
Knowing the sequence of proteins encoded by a gene can allow researchers to apply methods of finding similarity between protein sequences that provide more information than similarities or differences between proteins. DNA sequence.
The content above is for reference in 7.15A: Gene Families
Prepare the data
In this example, we will use data on human, chimpanzee and dog DNA, this example is based on the repo of author Nagesh Singh Chauhan so the data used for the three species above is also. Available in the DNA-Sequence-Machine-learning repo. The data includes groups of genes labeled as follows:
| Gene Family | Class label |
|---|---|
| G protein coupled receptors | 0 |
| Tyrosine kinase | first |
| Tyrosine phosphatase | 2 |
| Synthetase | 3 |
| Synthase | 4 |
| Ion channel | 5 |
| Transcription factor | 6 |
After downloading the above data, first as usual, we import the libraries we need to use:
1 2 3 4 5 6 | <span class="token keyword">import</span> numpy <span class="token keyword">as</span> np <span class="token keyword">import</span> pandas <span class="token keyword">as</span> pd <span class="token keyword">import</span> matplotlib <span class="token punctuation">.</span> pyplot <span class="token keyword">as</span> plt <span class="token keyword">from</span> functools <span class="token keyword">import</span> partial |
Next, with the pandas library we read the downloaded data as follows:
1 2 3 4 | humandf <span class="token operator">=</span> pd <span class="token punctuation">.</span> read_table <span class="token punctuation">(</span> <span class="token string">'data/human_data.txt'</span> <span class="token punctuation">)</span> chimpdf <span class="token operator">=</span> pd <span class="token punctuation">.</span> read_table <span class="token punctuation">(</span> <span class="token string">'data/chimp_data.txt'</span> <span class="token punctuation">)</span> dogdf <span class="token operator">=</span> pd <span class="token punctuation">.</span> read_table <span class="token punctuation">(</span> <span class="token string">'data/dog_data.txt'</span> <span class="token punctuation">)</span> |
Converts DNA sequence data into a k-mer characteristic matrix
The k-mer counting technique can be understood as the string into “words” with overlapping length. In this example, we will use k = 6. The following function is used to apply the k-mer technique:
1 2 3 | <span class="token keyword">def</span> <span class="token function">Kmers_funct</span> <span class="token punctuation">(</span> seq <span class="token punctuation">,</span> size <span class="token operator">=</span> <span class="token number">4</span> <span class="token punctuation">)</span> <span class="token punctuation">:</span> <span class="token keyword">return</span> <span class="token punctuation">[</span> seq <span class="token punctuation">[</span> x <span class="token punctuation">:</span> x <span class="token operator">+</span> size <span class="token punctuation">]</span> <span class="token punctuation">.</span> lower <span class="token punctuation">(</span> <span class="token punctuation">)</span> <span class="token keyword">for</span> x <span class="token keyword">in</span> <span class="token builtin">range</span> <span class="token punctuation">(</span> <span class="token builtin">len</span> <span class="token punctuation">(</span> seq <span class="token punctuation">)</span> <span class="token operator">-</span> size <span class="token operator">+</span> <span class="token number">1</span> <span class="token punctuation">)</span> <span class="token punctuation">]</span> |
Next we have the sentencize function to create k-mer “sentences”
1 2 3 4 5 6 7 8 9 10 | <span class="token keyword">def</span> <span class="token function">sentencize</span> <span class="token punctuation">(</span> df <span class="token punctuation">)</span> <span class="token punctuation">:</span> <span class="token triple-quoted-string string">'''Generate a df into kmers sentences and target vectors'''</span> seq_list <span class="token operator">=</span> df <span class="token punctuation">[</span> <span class="token string">'sequence'</span> <span class="token punctuation">]</span> <span class="token punctuation">.</span> tolist <span class="token punctuation">(</span> <span class="token punctuation">)</span> y_gt <span class="token operator">=</span> df <span class="token punctuation">[</span> <span class="token string">'class'</span> <span class="token punctuation">]</span> <span class="token punctuation">.</span> tolist <span class="token punctuation">(</span> <span class="token punctuation">)</span> <span class="token comment"># apply Kmer-counting to seq-list</span> kmers_list <span class="token operator">=</span> <span class="token builtin">map</span> <span class="token punctuation">(</span> partial <span class="token punctuation">(</span> Kmers_funct <span class="token punctuation">,</span> size <span class="token operator">=</span> <span class="token number">6</span> <span class="token punctuation">)</span> <span class="token punctuation">,</span> seq_list <span class="token punctuation">)</span> <span class="token comment">#k-mer words of length 6</span> sentences <span class="token operator">=</span> <span class="token punctuation">[</span> <span class="token string">' '</span> <span class="token punctuation">.</span> join <span class="token punctuation">(</span> seq <span class="token punctuation">)</span> <span class="token keyword">for</span> seq <span class="token keyword">in</span> kmers_list <span class="token punctuation">]</span> <span class="token keyword">return</span> sentences <span class="token punctuation">,</span> y_gt |
From there we obtain sents_human and y_human through the following statement:
1 2 | sents_human <span class="token punctuation">,</span> y_human <span class="token operator">=</span> sentencize <span class="token punctuation">(</span> humandf <span class="token punctuation">)</span> |
Continuously vectorize using BOW as well as dividing train and test:
1 2 3 4 5 6 7 | tf <span class="token operator">=</span> CountVectorizer <span class="token punctuation">(</span> ngram_range <span class="token operator">=</span> <span class="token punctuation">(</span> <span class="token number">4</span> <span class="token punctuation">,</span> <span class="token number">4</span> <span class="token punctuation">)</span> <span class="token punctuation">)</span> <span class="token comment"># 4-gram </span> x_human <span class="token operator">=</span> tf <span class="token punctuation">.</span> fit_transform <span class="token punctuation">(</span> sents_human <span class="token punctuation">)</span> <span class="token keyword">from</span> sklearn <span class="token punctuation">.</span> model_selection <span class="token keyword">import</span> train_test_split x_train <span class="token punctuation">,</span> x_test <span class="token punctuation">,</span> y_train <span class="token punctuation">,</span> y_test <span class="token operator">=</span> train_test_split <span class="token punctuation">(</span> x_human <span class="token punctuation">,</span> y_human <span class="token punctuation">,</span> test_size <span class="token operator">=</span> <span class="token number">0.2</span> <span class="token punctuation">,</span> random_state <span class="token operator">=</span> <span class="token number">42</span> <span class="token punctuation">)</span> |
Classification
Classification by Multinomial Naive Bayes
Initialize the model and train with the following command:
1 2 3 4 5 6 7 8 | <span class="token keyword">from</span> sklearn <span class="token punctuation">.</span> naive_bayes <span class="token keyword">import</span> MultinomialNB <span class="token comment">#create a model</span> clf_bayes <span class="token operator">=</span> MultinomialNB <span class="token punctuation">(</span> alpha <span class="token operator">=</span> <span class="token number">0.1</span> <span class="token punctuation">)</span> <span class="token comment">#train on data</span> clf_bayes <span class="token punctuation">.</span> fit <span class="token punctuation">(</span> x_train <span class="token punctuation">,</span> y_train <span class="token punctuation">)</span> |
Then using the trained model to guess on the test set, y_pred is y_pred
1 2 | y_pred <span class="token operator">=</span> clf_bayes <span class="token punctuation">.</span> predict <span class="token punctuation">(</span> x_test <span class="token punctuation">)</span> |
From that result, we can evaluate by the following popular methods:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 | <span class="token comment">#look at model performance: confusion matrix, accuracy, precision, recall, f1 score</span> <span class="token keyword">from</span> sklearn <span class="token punctuation">.</span> metrics <span class="token keyword">import</span> accuracy_score <span class="token punctuation">,</span> f1_score <span class="token punctuation">,</span> precision_score <span class="token punctuation">,</span> recall_score <span class="token keyword">print</span> <span class="token punctuation">(</span> <span class="token string">"Confusion matrix for predictions on human test DNA sequencen"</span> <span class="token punctuation">)</span> <span class="token keyword">print</span> <span class="token punctuation">(</span> pd <span class="token punctuation">.</span> crosstab <span class="token punctuation">(</span> pd <span class="token punctuation">.</span> Series <span class="token punctuation">(</span> y_test <span class="token punctuation">,</span> name <span class="token operator">=</span> <span class="token string">'Actual'</span> <span class="token punctuation">)</span> <span class="token punctuation">,</span> pd <span class="token punctuation">.</span> Series <span class="token punctuation">(</span> y_pred <span class="token punctuation">,</span> name <span class="token operator">=</span> <span class="token string">'Predicted'</span> <span class="token punctuation">)</span> <span class="token punctuation">)</span> <span class="token punctuation">)</span> <span class="token keyword">def</span> <span class="token function">calculate_metrics</span> <span class="token punctuation">(</span> y_test <span class="token punctuation">,</span> y_predicted <span class="token punctuation">)</span> <span class="token punctuation">:</span> accuracy <span class="token operator">=</span> accuracy_score <span class="token punctuation">(</span> y_test <span class="token punctuation">,</span> y_predicted <span class="token punctuation">)</span> precision <span class="token operator">=</span> precision_score <span class="token punctuation">(</span> y_test <span class="token punctuation">,</span> y_predicted <span class="token punctuation">,</span> average <span class="token operator">=</span> <span class="token string">'weighted'</span> <span class="token punctuation">)</span> recall <span class="token operator">=</span> recall_score <span class="token punctuation">(</span> y_test <span class="token punctuation">,</span> y_predicted <span class="token punctuation">,</span> average <span class="token operator">=</span> <span class="token string">'weighted'</span> <span class="token punctuation">)</span> f1 <span class="token operator">=</span> f1_score <span class="token punctuation">(</span> y_test <span class="token punctuation">,</span> y_predicted <span class="token punctuation">,</span> average <span class="token operator">=</span> <span class="token string">'weighted'</span> <span class="token punctuation">)</span> <span class="token keyword">print</span> <span class="token punctuation">(</span> <span class="token string">"accuracy = %.3f nprecision = %.3f nrecall = %.3f nf1 = %.3f"</span> <span class="token operator">%</span> <span class="token punctuation">(</span> accuracy <span class="token punctuation">,</span> precision <span class="token punctuation">,</span> recall <span class="token punctuation">,</span> f1 <span class="token punctuation">)</span> <span class="token punctuation">)</span> calculate_metrics <span class="token punctuation">(</span> y_test <span class="token punctuation">,</span> y_pred <span class="token punctuation">)</span> |
From there we obtained the following results
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 | Confusion matrix <span class="token keyword">for</span> predictions on human <span class="token builtin class-name">test</span> DNA sequence Predicted <span class="token number">0</span> <span class="token number">1</span> <span class="token number">2</span> <span class="token number">3</span> <span class="token number">4</span> <span class="token number">5</span> <span class="token number">6</span> Actual <span class="token number">0</span> <span class="token number">99</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">1</span> <span class="token number">0</span> <span class="token number">2</span> <span class="token number">1</span> <span class="token number">0</span> <span class="token number">104</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">2</span> <span class="token number">2</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">78</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">3</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">124</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">1</span> <span class="token number">4</span> <span class="token number">1</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">143</span> <span class="token number">0</span> <span class="token number">5</span> <span class="token number">5</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">51</span> <span class="token number">0</span> <span class="token number">6</span> <span class="token number">1</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">1</span> <span class="token number">0</span> <span class="token number">0</span> <span class="token number">263</span> accuracy <span class="token operator">=</span> <span class="token number">0.984</span> precision <span class="token operator">=</span> <span class="token number">0.984</span> recall <span class="token operator">=</span> <span class="token number">0.984</span> f1 <span class="token operator">=</span> <span class="token number">0.984</span> |
Machine title vector
The following code is used to train as well as evaluate the results of the model
1 2 3 4 5 6 7 8 9 10 | <span class="token keyword">from</span> sklearn <span class="token keyword">import</span> svm clf_svm <span class="token operator">=</span> svm <span class="token punctuation">.</span> SVC <span class="token punctuation">(</span> C <span class="token operator">=</span> <span class="token number">100</span> <span class="token punctuation">,</span> gamma <span class="token operator">=</span> <span class="token number">0.001</span> <span class="token punctuation">,</span> kernel <span class="token operator">=</span> <span class="token string">'rbf'</span> <span class="token punctuation">)</span> <span class="token comment">#train</span> clf_svm <span class="token punctuation">.</span> fit <span class="token punctuation">(</span> x_train <span class="token punctuation">,</span> y_train <span class="token punctuation">)</span> <span class="token comment">#predict and eval</span> calculate_metrics <span class="token punctuation">(</span> y_test <span class="token punctuation">,</span> clf_svm <span class="token punctuation">.</span> predict <span class="token punctuation">(</span> x_test <span class="token punctuation">)</span> <span class="token punctuation">)</span> |
The results are as follows:
1 2 3 4 5 | accuracy <span class="token operator">=</span> <span class="token number">0.893</span> precision <span class="token operator">=</span> <span class="token number">0.921</span> recall <span class="token operator">=</span> <span class="token number">0.893</span> f1 <span class="token operator">=</span> <span class="token number">0.896</span> |
Predictions from DNA sequence data of other species
Similar to processing human genome data, we proceed with DNA data of other species as follows:
1 2 3 4 5 6 | sents_chimp <span class="token punctuation">,</span> y_chimp <span class="token operator">=</span> sentencize <span class="token punctuation">(</span> chimpdf <span class="token punctuation">)</span> sents_dog <span class="token punctuation">,</span> y_dog <span class="token operator">=</span> sentencize <span class="token punctuation">(</span> dogdf <span class="token punctuation">)</span> x_chimp <span class="token operator">=</span> tf <span class="token punctuation">.</span> transform <span class="token punctuation">(</span> sents_chimp <span class="token punctuation">)</span> x_dog <span class="token operator">=</span> tf <span class="token punctuation">.</span> transform <span class="token punctuation">(</span> sents_dog <span class="token punctuation">)</span> |
Next, define the evaluate_classifier function as follows:
1 2 3 4 5 6 7 8 9 10 11 | <span class="token keyword">def</span> <span class="token function">evaluate_classifier</span> <span class="token punctuation">(</span> model <span class="token punctuation">)</span> <span class="token punctuation">:</span> <span class="token comment">#predict</span> y_pred_chimp <span class="token operator">=</span> model <span class="token punctuation">.</span> predict <span class="token punctuation">(</span> x_chimp <span class="token punctuation">)</span> y_pred_dog <span class="token operator">=</span> model <span class="token punctuation">.</span> predict <span class="token punctuation">(</span> x_dog <span class="token punctuation">)</span> <span class="token comment">#eval</span> <span class="token keyword">print</span> <span class="token punctuation">(</span> <span class="token string">"Performance on Chimpanzee test DNA"</span> <span class="token punctuation">)</span> calculate_metrics <span class="token punctuation">(</span> y_chimp <span class="token punctuation">,</span> y_pred_chimp <span class="token punctuation">)</span> <span class="token keyword">print</span> <span class="token punctuation">(</span> <span class="token string">"----nPerformance on Dog test DNA"</span> <span class="token punctuation">)</span> calculate_metrics <span class="token punctuation">(</span> y_dog <span class="token punctuation">,</span> y_pred_dog <span class="token punctuation">)</span> |
From there using the evaluate_classifier function with two models trained with our human DNA data obtained:
- Using Multinomial Naive Bayes model
- Prediction on chimpanzee DNA
- accuracy = 0.993
- precision = 0.994
- recall = 0.993
- f1 = 0.993
- Prediction on dog DNA data
- accuracy = 0.926
- precision = 0.934
- recall = 0.926
- f1 = 0.925
- Prediction on chimpanzee DNA
- Use the model vector machine title:
- Prediction on chimpanzee DNA
- accuracy = 0.968
- precision = 0.971
- recall = 0.968
- f1 = 0.969
- Prediction on dog DNA data
- accuracy = 0.493
- precision = 0.803
- recall = 0.493
- f1 = 0.436
- Prediction on chimpanzee DNA
From the above results it can be seen that the SVM model shows that humans and chimpanzees are much more closely related than dogs since the results are worthwhile using a predictive model with low dog DNA data. much more when using chimpanzee DNA guessing.
Conclude
Bioinformatics – research that integrates high-flux bio data and statistical modeling through computationally intensive computing, has attracted great attention recently and DNA sequencing is one of the Its core problem. The vast amount of information gained from sequencing has given us a deeper understanding and basic understanding of the organism. The above article briefly covers the basics of DNA, introduces some libraries used to visualize and process data, and finally a small example of the application of scientific methods. computers that are more specific are machine learning methods for processing and extracting some insights into the DNA sequence data of some species.